Cross Validation#
[1]:
%load_ext autoreload
%autoreload 2
from tdm.raw.breast_mibi import read_single_cell_df
from tdm.model.selection import cross_validation2, plot_cross_validation_result
from tdm.cell_types import FIBROBLAST, MACROPHAGE, TUMOR, ENDOTHELIAL
single_cell_df = read_single_cell_df()
Define the settings to compare by constructing a dictionary that maps a setting name, to kwargs for the model and feature transforms:
[13]:
setting_dicts = {
'degree 2': {
'model_kwargs': {}, # see: LogisticRegressionModel for more options such as regularization
'polynomial_dataset_kwargs': {'degree': 2} # see: PolynomialDataset for more feature transformations
},
'degree 1': {
'model_kwargs': {},
'polynomial_dataset_kwargs': {'degree': 1},
}
}
Run the cross validation (note: partitions the dataset at the level of tissues, not cells)
[14]:
res = cross_validation2(
single_cell_df,
model_dicts=setting_dicts,
cell_types_to_model=[FIBROBLAST, MACROPHAGE],
allowed_neighbor_types=[FIBROBLAST, MACROPHAGE, TUMOR, ENDOTHELIAL],
neighborhood_mode='extrapolate',
n_splits=3
)
100%|██████████| 3/3 [00:03<00:00, 1.29s/it]
Plot the result:
[15]:
plot_cross_validation_result(res)
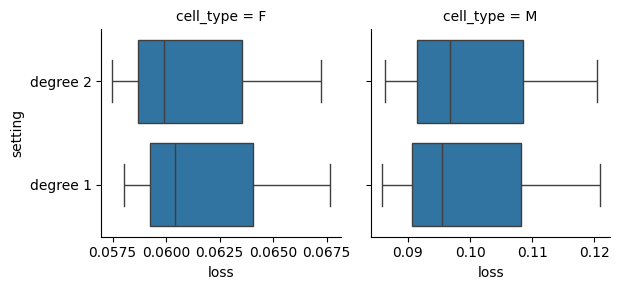
There are no major differences over the settings above, for simplicity we can take the best performing model on average over all cell types:
[16]:
res.groupby('setting').loss.mean().sort_values()
[16]:
setting
degree 2 0.081324
degree 1 0.081365
Name: loss, dtype: float64